Хочу ещё раз напомнить, что
диагноз может поставить только врач при осмотре и после ряда дополнительных обследований!
СИНДРОМ МАРФАНА: Что такое синдром Марфана? + Проявления синдрома Марфана + Причина синдрома Марфана
| РЕГИСТРИРУЙТЕСЬ на нашем НОВОМ форуме по адресу: КРУГ ДРУЗЕЙ |
Привет, Гость! Войдите или зарегистрируйтесь.
Вы здесь » В ГОСТЯХ У ОЛЬГИ-АЛЁНУШКИ » РАЗНОЕ + ЭТО ИНТЕРЕСНО » Врождённые и генетические заболевания

Хочу ещё раз напомнить, что
диагноз может поставить только врач при осмотре и после ряда дополнительных обследований!
СИНДРОМ МАРФАНА: Что такое синдром Марфана? + Проявления синдрома Марфана + Причина синдрома Марфана
Синдром Марфана
Что такое синдром Марфана?
Синдром Марфана – наследственное заболевание, характеризующееся поражением соединительной ткани. Соединительная ткань входит в состав разных органов организма, обеспечивает поддержание структуры тела. При синдроме Марфана соединительная ткань имеет дефекты, что может проявляться патологиями разных систем организма, включая скелет, глаза, кровеносные сосуды, нервную систему, кожу, легкие и др.
Синдром Марфана встречается у людей всех рас и представителей разных этнических групп, как у мужчин, так и у женщин.
Проявления синдрома Марфана
Синдром Марфана может у разных людей проявляться по-разному. У некоторых людей наблюдаются легкие проявления болезни, в то время как у других - тяжелые. В большинстве случаев симптомы прогрессируют с возрастом человека. Синдром Марфана может влиять на разные системы организма:
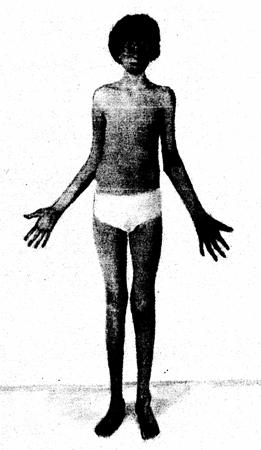
* Скелет - человек с синдромом Марфана обычно очень высокий и худой. В связи с тем, что синдром Марфана сопровождается удлинением костей скелета: туловища, рук, ног, пальцев рук и ног, они могут быть непропорционально длинные. Человек с синдромом Марфана часто имеет длинное, узкое лицо, и его верхняя губа может быть изогнута, причиной чего являются зубы. Другие скелетные аномалии включают изменение грудины (грудной кости), которая или выступает, или зигзагообразной формы, искривление спины (сколиоз) и плоскостопие.
* Глаза – у более, чем половины всех людей с синдромом Марфана отмечается смещение одного из двух хрусталиков глаз. Хрусталики глаз могут быть незначительно выше, чем нормальные и смещаться в сторону. Смещение может быть минимальным или резко выраженным и очевидным. Отслоение сетчатки – это серьёзное осложнение. Многие люди с синдромом Марфана близоруки и у них может развиться глаукома (высокое давление внутри глаза) или катаракта.
* Сердце и кровеносные сосуды (сердечно-сосудистая система) – большинство людей с синдромом Марфана имеют аномалии, связанные с сердцем и кровеносными сосудами. Так как имеется дефект соединительной ткани, стенка аорты (большая артерия, которая несет кровь из сердца к телу) может быть ослаблена и растягиваться, этот процесс называют расширением аорты. Расширение аорты - увеличивает риск разрыва аорты, вследствие чего возникает серьёзная проблема с сердцем или иногда внезапная смерть. Иногда могут развиваться дефекты сердечных клапанов. В большинстве случаев, определенные клапаны могут создавать «сердечный шум», который может услышать доктор с помощью стетоскопа. Небольшие нарушения в работе сердца могут не вызывать симптомов, но большие могут стать причиной затруднения дыхания, усталости и сильного сердцебиения (очень быстрого или неправильного сердечного ритма).
* Нервная система – головной и спинной мозг окружены жидкостью; вокруг имеется оболочка, которая называется твердой мозговой оболочкой, ее составляет соединительная ткань. Когда люди с синдромом Марфана стареют, твердая мозговая оболочка часто слабеет и вытягивается. Это называется дуральная эктазия. Эти изменения могут стать причиной легкого дискомфорта или могут привести к боли в брюшной полости или боли, неподвижности или слабости ног.
* Кожа - у большинства людей с синдромом Марфана развивается растягивание кожи, даже без изменения массы тела. Это может появиться в любом возрасте и не представлять опасности. Тем не менее, у людей с синдромом Марфана увеличивается риск развития брюшной или паховой грыжи, при которой развивается выпуклость, которая содержит в себе часть кишки.
* Легкие – хотя аномалии соединительной ткани делают маленькие воздушные мешочки в легких менее эластичным, люди с синдромом Марфана обычно не испытывают существенных проблем с их легкими. Если, тем не менее, эти маленькие воздушные мешочки удлиняются или распухают, то может увеличиться риск спадения легких. Редко у людей с синдромом Марфана могут возникать нарушения дыхания во время сна, как храп или синдром апноэ (нарушение сна возникает в краткий период, когда дыхание останавливается).
Причина синдрома Марфана
Синдром Марфана развивается вследствие дефекта (изменения) в гене, который определяет структуру фибрина, который играет огромную роль в соединительной ткани. Человек с синдромом Марфана рожден с нарушением, даже если это не было установлено в течение жизни. Хотя каждый человек с синдромом Марфана имеет дефект определенного гена, изменения специфичны для каждой семьи и не каждый может испытывать подобные симптомы в одинаковой степени. Это называют подразумевает, что дефектный ген проявляется у различных людей в различной степени.
Дефектный ген может быть унаследован: ребенок имеет 50% вероятность унаследовать заболевание от родителя с синдромом Марфана. Иногда новый генный дефект появляется во время соединения сперматозоида и яйцеклетки, здоровые родители имеют один шанс из 10,000, родить ребенка с синдромом Марфана. Возможно, в 25% случаев происходит самопроизвольное изменение во время оплодотворения.
Синдром Марфана
Как устанавливается синдром Марфана?
Нет специальных лабораторных анализов таких, как анализ крови или биопсия кожи, чтобы диагностировать синдром Марфана. Доктор или генетик (врач со специальными знаниями о наследственных заболеваниях) полагается на обследование и историю болезни, включая информацию о членах семьи, которые могут иметь нарушение или у кого была необъяснимая смерть от сердечного приступа, на полное физическое обследование, включая осмотр скелета для оценки пропорциональности размера руки/ноги к размеру туловища, на обследование зрения, включая осмотр «щелевой лампой», обследование сердца, такое как ультразвуковое исследование (анализ, в котором используются ультразвуковые волны, чтобы проверить сердце и аорту).
Врач может поставить диагноз синдрома Марфана, если в семье пациента у кого-то есть эта болезнь и есть особые проблемы. У пациента без соответствующей истории болезни должны быть затронуты три системы организма, чтобы был поставлен диагноз. Кроме того, две из систем должны иметь симптомы, которые сравнительно специфичны для синдрома Марфана. В некоторых случаях, может быть полезен генетический анализ, но некоторые анализы часто требуют времени и могут не содержать дополнительной полезной информации. Члены семьи человека, у которого диагностировали синдром Марфана, не должны предполагать, что они повлияли на это, если нет информации о том, не возникало ли нарушение в предыдущем поколении семьи.
Какие врачи лечат синдром Марфана?
В связи с тем, что ряд систем организма может быть подвержен воздействию, человек с синдромом Марфана должен наблюдаться у различных врачей. Терапевт или педиатр может направить пациента к таким специалистам, как кардиолог (врач, который специализируется на пороках сердца), ортопед (врач - специалист по костям), или офтальмолог (врач - специалист по заболеваниям глаз), если нужно. Некоторые люди с синдромом Марфана лечатся у генетика.
Какие виды лечения доступны при синдроме Марфана?
Нет лечения для синдрома Марфана. Совершенствуясь, ученые имеют возможность определить и изменить специфику гена, который ответственен за нарушение при рождении. Тем не менее, цель лечения - предупреждение и лечение осложнений. Соответствующие специалисты будут совершенствовать индивидуальную программу лечения; подход врача зависит от того, какие системы затронуты.
* Скелет - Ежегодные обследования очень важны, чтобы определить изменения позвоночника или грудины. Это очень важно во время быстрого роста в период юности. Серьезные деформации могут нарушить функционирование сердца и легких. В некоторых случаях, могут быть рекомендованы ортопедический корсет или операция, чтобы снизить вред и искривление.
* Глаза - регулярное обследование глаз очень важно, чтобы приостановить и скорректировать проблемы со зрением, связанные с синдромом Марфана. В большинстве случаев, очки или контактные линзы могут исправить проблему, хотя в некоторых случаях может быть необходима хирургическая операция.
* Сердечно-сосудистая система - регулярное обследование и кардиограммы помогут врачу оценить размер аорты и работу сердца. На ранней стадии проблема устанавливается и лечится, иначе возможен риск опасных для жизни осложнений. Другие люди с проблемами сердца обращаются в кабинет неотложной помощи, если они испытывают боль в грудной клетке, спине или в животе. Некоторые проблемы с сердечными клапанами могут быть разрешены с помощью медикаментов, таких как бета-адреноблокаторы. В других случаях, хирургическое вмешательство по замене клапана или восстановлению аорты может быть необходимо. После операции на сердце, необходим хороший уход, чтобы предотвратить эндокардит. Стоматологи должны помнить о риске; они, вероятно, посоветуют, пациенту принимать соответствующие медикаменты, прежде чем начнут лечение.
* Нервная система - если развивается дуральная эктазия, то лечение может помочь минимизировать боль.
* Легкие - Особенно важно, чтобы люди с синдромом Марфана не курили, так как у них возрастает риск заболеваний легких. При появлении проблем с дыханием нужно обратиться к врачу.
Беременность может оказать влияние на организм, редко на сердце. Беременность должна протекать под наблюдением акушеров-гинекологов и других специалистов, беременность при синдроме Марфана должна рассматривается как опасное состояние. При питании важна сбалансированная диета, поддержание здорового образа жизни, но без витаминов или добавок, которые малоэффективны и не влияют на синдром Марфана.
Синдром Марфана
Эмоциональные и психологические проявления синдрома Марфана
Наличие диагноза и неумение жить с генетическим нарушением может быть причиной социального, эмоционального и финансового стресса. Это требует осмысления будущих планов и стиля жизни. Человек, уже вполне взрослый, когда ему диагностируют синдром Марфана, может чувствовать себя сердитым или испуганным.
Могут возникать вопросы о том, как влияет нарушение на будущее поколение или о том, какие физические, эмоциональные и финансовые последствия могут быть.
Родители, брат или сестра человека, у которого диагностировали синдром Марфана, могут чувствовать себя печально, раздраженно и винить себя. Очень важно для родителей знать, что они не являются причиной того, что ген фибрилина видоизменяется. Родители могут поговорить о генетическом наследовании или о том, есть ли риск для будущих детей. Некоторые люди с синдромом Марфана должны ограничивать свою физическую деятельность. Это может требовать переосмысления образа жизни.
Для детей и взрослых необходима медицинская помощь, точная информация и социальная поддержка - это главное в жизни с болезнью. Генетическая консультация может быть полезной в осмыслении болезни и ее влиянии на будущее поколение.
Какое будущее у человека с синдромом Марфана?
Пока синдром Марфана - это постоянное нарушение, но медицинские методы улучшаются с каждым годом. Раннее обнаружение диагноза и прогресс медицинских технологий улучшает качество жизни людей с синдромом Марфана и увеличивает продолжительность их жизни. Кроме того, раннее обнаружение факторов риска (таких, как расширение аорты) позволяет врачу вмешаться и предотвратить, или отсрочить осложнения. Исследования дают людям надежду на будущее. При раннем установлении диагноза и соответствующим лечением жизнь человека с синдромом Марфана становится такой же, как и у нормального человека
Какие исследования помогают людям с синдромом Марфана?
Ученые исследуют синдром Марфана с точки зрения различных перспектив. Одно исследование пытается понять, как возникает генетический дефект или изменение. Как развивается соединительная ткань и как функционирует в теле? Почему у людей с синдромом Марфана возникают разные изменения? Ученые ищут ответы на многие вопросы при изучении гена и семей, затронутых болезнью. Подопытные мыши, которые имеют изменение в гене фибрилина, могут помочь ученым лучше понять нарушение. Исследования на животных - "подготовительный" этап генной терапии, и они идут полным ходом.
Другие ученые фокусируют свое внимание на лечении некоторых осложнений, которые возникают у людей с синдромом Марфана. Клинические исследования могут установить неэффективность некоторых медикаментов при предупреждении или уменьшении проблем с аортой. Исследователи работают, чтобы усовершенствовать новые методы хирургического лечения, чтобы улучшить состояние здоровья людей с синдромом Марфана.
Очень информативная ссылка Синдром Марфана
Статья из медицинской энциклопедии СИНДРОМ МАРФАНА
Синдром Марфана в практике терапевта и семейного врача: диагностика, тактика ведения, лечение, беременность и роды
Среди всех наследственных заболеваний соединительной ткани наибольший интерес для терапевтов и врачей общей практики представляет синдром Марфана, так как продолжительность жизни этих больных ограничена 30–40 годами и у одного пациента может быть столько проблем со здоровьем, сколько специалистов в поликлинике. Поскольку заболевание имеет заведомо серьезный прогноз для жизни и трудоспособности пациентов, установление диагноза накладывает особую ответственность на врача при первой встрече с больным.
В 1896 году французский профессор–педиатр Антонио Марфан впервые представил клиническое наблюдение 5–летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета. Девочка умерла в юном возрасте, вероятно, от туберкулеза. Внешний габитус Габриель и подобных пациентов с тех пор стали именовать синдромом Марфана. Как позднее выяснилось, в действительности Габриель страдала врожденной контрактурной арахнодактилией. Через 20 лет были описаны первые фенокопии марфаноподобных синдромов, в частности, синдрома эктопии хрусталиков с аутосомно–доминантным наследованием, еще через 30 лет – синдрома дилатации и расслоения аорты, пролапса митрального клапана, эктазии твердой мозговой оболочки.
Weve H. впервые предположил, что причиной синдрома Марфана является дефект мезодермы, а известный американский генетик McKusick в расписании наследственных болезней человека «On–Line Mendelian Inheritance In Man» (OMIM) открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани. Фенотип синдрома характеризуется некоторой протяженностью, начиная от легких, «мягких» проявлений соединительнотканой дисплазии, встречающихся также и в общей популяции, до случаев с угрожающими жизни системными расстройствами.
Основной документ, на котором базировался диагноз синдрома Марфана, был представлен в 1986 году – это так называемая Berlin Nosology. Среди критериев Берлинской Нозологии превыше всего ставились достижения молекулярной генетики. Однако установленная локализация гена синдрома Марфана в аутосоме 15q21, кодирующего микрофибриллярный белок фибриллин–1, не является единственной и характерной исключительно для синдрома Марфана. Мутация в гене родственного протеина – фибриллина–2 также ведет к клиническим проявлениям марфаноидного габитуса. Нозологические формы с фенотипом «Марфана», такие как контрактурная арахнодактилия и семейный пролапс митрального клапана – MASS–фенотип, имели мутации в тех же генах. Большинство ошибочных диагнозов у родственников больных, как оказалось, связаны с переоценкой значимости молекулярно–генетических исследований, так как в случае их позитивности в семейной истории болезни приводили к предвзятости диагноза у других членов семьи. Только совместные молекулярно–генетические и клинические исследования имеют достаточные основания для создания полноценных диагностических критериев.
Современные критерии диагноза синдрома Марфана разработаны в 1996 году совместными усилиями генетиков и клиницистов и предлагаются к широкому использованию врачами всех специальностей.
«Большим» критерий считается вследствие его большей специфичности, так как он редко встречается при других состояниях и в общей популяции. В целом диагностическое решение должно приниматься на основании больших критериев болезни. Важно отличать «большой критерий», имеющийся в системе органов и определяющий данное заболевание, от «системы органов, вовлеченной в процесс соединительнотканой дисплазии».
Диагностические критерии патологии скелета
Большие критерии. «Большим критерием» патологии скелета считается наличие не менее 4 из следующих признаков:
– килевидная деформация грудной клетки или воронкообразная деформация грудной клетки больших степеней, подлежащая оперативному лечению;
– уменьшение верхнего сегмента тела (рост сидя) по отношению к нижнему или если размах рук превышает рост на 5%;
– положительные тесты запястья и большого пальца (см. ниже);
– сколиоз более 20° или спондилолистез;
– невозможность полного разгибания локтевых суставов (угол < 170°);
– медиальное смещение внутренних лодыжек в результате продольного плоскостопия;
– протрузия вертлужной впадины любой степени (при рентгенографии).
Малые критерии:
– воронкообразная деформация грудной клетки умеренной степени;
– гипермобильность суставов;
– высокое аркообразное нёбо со «скученностью» зубов;
– аномалии черепа и лица (долихоцефалия, гипоплазия скул, эндофтальмия – глубоко посаженные глаза, ретрогнатия, косо опущенные складки век).
Патология скелета для верификации диагноза «синдром Марфана» должна быть представлена двумя большими критериями (при наличии всех признаков) или одним большим критерием (4 признака) и двумя малыми критериями.
Комментарии. Многие скелетные аномалии часто встречаются в популяции, однако комбинация определенных вышеописанных дефектов является высокоспецифичной для диагноза. Например, гипермобильность суставов – один из примеров высокой распространенности среди населения, поэтому самостоятельная значимость этого признака очень мала и он не включен в большие диагностические критерии. Напротив, такой признак, как врожденные суставные контрактуры, редки в общей популяции, но не часто встречаются и при синдроме Марфана (характерны контрактуры локтевых суставов). В случае выраженной контрактуры суставов и снижения суставной мобильности в сочетании с другими скелетными аномалиями необходимо дифференцировать синдром Марфана с врожденной контрактурной арахнодактилией.
Для деформаций грудной клетки при синдроме Марфана более характерна грудина, выступающая вперед и смещение кпереди левого реберно–хондрального соединения, что придает грудной клетке асимметричность.
Соотношение роста и размаха рук оценивают с учетом антропометрических коэффициентов, полученных Gordon С.С. с соавторами при антропометрии личного состава американской армии в 1988 году. Отношение верхний/нижний сегменты изучено у лиц всех возрастов McKusick, а позднее другими.
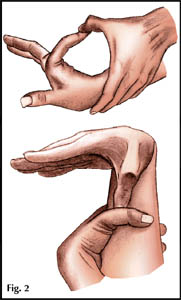
Тест запястья заключается в обхвате запястья большим пальцем и мизинцем; их терминальные фаланги при этом накладываются друг на друга. Тест большого пальца заключается в фиксации его поперек ладони без дополнительной помощи: положительным считается, когда ногтевая фаланга большого пальца выходит за ульнарный край ладони.
Сколиозы различных степеней, обычно грудного отдела, выпуклостью вправо, имеют место, по крайней мере, у 60% пациентов. Аномалии позвоночника в сагиттальной плоскости, такие как выпрямление кифоза или гиперкифоз также заслуживают внимания. Спондилолистез встречается в 6% случаев.
Для подтверждения диагноза синдрома Марфана необходимо выявить протрузию вертлужной впадины. Для этого рекомендуют рентгенографию, либо методы, уменьшающие или исключающие лучевую нагрузку – компьютерную томографию или магнитно–резонансную томографию.
Диагностические критерии патологии зрения
Большие критерии:
– эктопия хрусталиков.
Малые критерии:
– патологически плоская роговица (выявленная с помощью кератометрии);
– увеличенная длина глазного яблока (измеренная ультрасонографически);
– гипоплазия радужной оболочки или гипоплазия цилиарной мышцы, ведущие к ухудшению миоза и аккомодации.
Для диагноза значимо вовлечение окулярной системы, о чем говорит наличие 1 большого или, по крайней мере, 2 малых критериев.
Комментарии. Адекватная оценка эктопии хрусталиков возможна при полной дилатации зрачка и исследовании в щелевой лампе. Дислокация бывает одно– или двусторонняя и в любом направлении, хотя чаще находят смещение вверх. Иридодонез (трепетание радужной оболочки) является вторичным к эктопии хрусталика, поэтому не может рассматриваться, как отличительный признак при синдроме Марфана.
Радиус кривизны роговицы должен быть изучен кератометрически. Степень уплощения роговицы положительно коррелирует с эктопией хрусталиков. Мегалокорнеа изредка обнаруживается при синдроме Марфана, но не может считаться даже малым критерием диагностики.
Длина оси глазного яблока при синдроме Марфана обычно увеличена (нормальная длина у взрослых < 23,5 мм). Увеличенная длина глазного яблока ведет к миопии и является предрасполагающим фактором отслойки сетчатки (последняя не считается самостоятельным критерием).
Гипоплазия цилиарной мышцы обнаруживается только в сочетании с гипоплазией радужной оболочки, поэтому наличие одной или обеих аномалий учитывается, как один малый критерий диагноза. Некоторые эксперты считают развитие ранней катаракты и открытоугольную глаукому типичными признаками синдрома Марфана. Однако эти признаки нуждаются в дальнейшей переоценке для включения их в категорию малых критериев.
Диагностические критерии патологии сердечно–сосудистой системы
Большие критерии:
– дилатация восходящей аорты с или без аортальной регургитации и вовлечением, по крайней мере, синусов Вальсальвы;
– расслоение восходящей аорты.
Малые критерии:
– пролапс митрального клапана с или без митральной регургитации;
– дилатация легочного ствола в отсутствие клапанного или подклапанного легочного стеноза или каких–либо других очевидных причин в возрасте до 40 лет;
– кальцификация митрального кольца в возрасте до 40 лет;
– дилатация или расслоение нисходящей грудной или брюшной аорты в возрасте до 50 лет.
Для оценки поражения кардиоваскулярной системы должен присутствовать большой критерий или один малый критерий.
Комментарии. Дилатация корня аорты диагностируется, если максимальный размер на уровне синусов Вальсальвы при эхокардиографии превышает нормальные значения для данного возраста и площади поверхности тела. Расслоение аорты документируется контрастной ангиографией, чреспищеводной эхокардиографией, компьютерной томографией (КТ) или магнитно–резонансной томографией (МРТ). Размеры легочного ствола принято оценивать по номограммам для аорты при эхокардиографии, КТ или МРТ. Убедительным критерием диагностики пролапса митрального клапана является позднесистолическое провисание створки более 2 мм в М–режиме эхокардиографии или парусящая в левое предсердие створка при секторальной эхокардиографии по длинной оси.
Диагностические критерии поражения бронхолегочной системы
Большие критерии отсутствуют.
Малые критерии:
– спонтанный пневмоторакс;
– апикальные буллы, обнаруженные при рентгенографии.
О вовлечении бронхолегочной системы в процесс судят по наличию одного из малых критериев.
Диагностические критерии патологии кожи и мягких тканей
Большие критерии отсутствуют.
Малые критерии:
– атрофические стрии (стрии вытяжения), не связанные с изменениями массы тела, беременностью или повторяющимися стрессами;
– рецидивирующая или оперированная грыжа.
О заинтересованности кожного и подлежащих покровов судят по наличию одного из малых критериев.
Комментарии. Стрии при синдроме Марфана локализованы преимущественно на плечах, в нижней или средней части спины, на бедрах. Стрии также встречаются при MASS–фенотипе и в общей популяции в определенных ситуациях (например, у женщин во время и после беременности).
Диагностический критерий поражения твердой мозговой оболочки
Большие критерии
– пояснично–крестцовая эктазия твердой мозговой оболочки при КТ или МРТ.
Малые критерии отсутствуют.
О вовлечении в процесс твердой мозговой оболочки свидетельствует большой критерий.
Комментарии. Адекватная оценка эктазии твердой мозговой оболочки осуществляется при КТ или МРТ пояснично–крестцового региона (обзор L5–S1). Эктазия твердой мозговой оболочки описывается, если:
– имеется расширение позвоночного канала вместе со спинным мозгом, ближе к нижнепоясничному или крестцовому отделам;
– истончение замыкательной пластинки позвонков и расширение отверстий нервных корешков;
– передняя мозговая грыжа.
Эктазию твердой мозговой оболочки относят к большим критериям диагноза, поскольку ее распространение при синдроме Марфана предположительно более 40%, а при других болезнях соединительной ткани этот признак практически не встречается. Если появляются серьезные изменения цереброспинальной жидкости, то это может привести к прогрессированию дилатации твердой мозговой оболочки и последующему эрозированию костей позвоночника. Эктазия твердой мозговой оболочки у детей встречается реже.
Диагностические критерии семейного анамнеза
Большие критерии:
– родители, дети или сибсы (братья, сестры), у которых имеются приведенные выше диагностические критерии;
– наличие мутации в гене белка фибриллина–1 (определена, как причина синдрома Марфана);
– наличие гаплотипа, подобного дефекту фибриллина–1, наследуемого потомству и однозначно ассоциированного с диагнозом синдрома Марфана в семьях.
Малые критерии отсутствуют.
Наличие одного из больших критериев семейного анамнеза свидетельствует в пользу диагноза.
Комментарии. Когда гаплотип был описан, он стал использоваться в диагностике не только у родственников 1 степени родства, но и у родственников 2 степени родства и более дальних. Невозможность определения мутации в гене фибриллина–1 или молекулярной аномалии самого фибриллина–1 не исключает у пациента синдром Марфана, если у него имеются клинические критерии.
Диагностика спорадического случая синдрома Марфана
· Если семейного анамнеза нет, то необходимо, по крайней мере, 2 больших критерия в разных органах и системах и вовлечение третьей системы.
· Если выявлена мутация, как причина синдрома Марфана, то клинически достаточно одного большого критерия и вовлечения второй системы органов.
Диагностика случаев у родственников больных
· Наличие большого критерия в семейном анамнезе и одного большого клинического критерия, а также вовлечения еще одной системы органов.
Комментарии. Невозможно диагностировать все нозологические подгруппы с марфаноидным фенотипом на основании этих исследований. Например, для исключения гомоцистинурии должен быть проведен анализ крови или мочи на содержание гомоцистина в отсутствие лечения пиридоксином.
Синдромы с марфаноподобным фенотипом
Синдром Марфана входит под номером 154700 в систему табуляции МакКьюсика – ОMIM. Перечисленные ниже состояния внешне очень напоминают синдром Марфана, но значительно отличаются по висцеральным поражениям, осложнениям и прогнозу.
1. Врожденная контрактурная арахнодактилия (ОMIM:121050),
2. Семейная грудная аневризма аорты (ОMIM:132900); в прошлом это состояние именовали Эрдхеймовский некроз средней оболочки аорты,
3. Семейное расслоение аорты (ОMIM:132900),
4. Семейная эктопия хрусталика (ОMIM:129600),
5. Семейный марфаноидный габитус (возможно, ОMIM:154705).
Рекомендации по диагнозу: большой критерий поражения скелета должен присутствовать, по крайней мере, у двух родственников. Полисистемное вовлечение органов может не выявляться, но не исключено. Если бы даже МакКьюсик не дифференцировал семейные случаи аневризмы аорты и расслоения аорты, практика показала бы, что члены семей с расслоением аорты не имели даже минимальной дилатации восходящей аорты, предшествующей расслоению.
6. MASS–фенотип: миопия, пролапс митрального клапана, умеренное расширение аорты (не более чем 2 стандартных отклонения), патология кожи (стрии) и скелета («малые критерии» синдрома Марфана).
Рекомендации по диагнозу: по крайней мере, 2 или даже 3 системы органов могут быть вовлечены в процесс.
7. Семейный синдром пролапса митрального клапана
Рекомендации по диагнозу: пролапс митрального клапана передается по аутосомно–доминантному типу; при этом могут отмечаться «мягкие» скелетные проявления, но они недостаточны для диагностики MASS–фенотипа. McKusick не дифференцирует эти два состояния: MASS–фенотип и семейный синдром пролапса митрального клапана находятся в одной рубрике ОMIM с индексом 109730.
8. Синдром Стиклера (наследственная артроофтальмопатия (ОMIM: 108300).
Рекомендации по диагнозу: это мультисистемное заболевание с поражением глаз, краниофасциальными дефектами и вовлечением, по меньшей мере, еще одной системы органов. Типичными находками могут быть: высокая миопия, дегенерация стекловидного тела и сетчатки, отслойка сетчатки, глухота; артропатия; умеренная спондилоэпифизарная дисплазия, изредка выявляющаяся на первых годах жизни; гипермобильность суставов; гипоплазия средней части лица; микрогнатия; аркообразное нёбо, пролапс митрального клапана. Включает синдром Вейссенбахера–Цвеймюллера (Weissenbacher–Zweymuller syndrome, ОMIM: 277610).
9. Синдром Шпринтзена–Гольберга (ОМIМ: 182212).
Рекомендации по диагнозу: вместе со скелетными аномалиями, напоминающими синдром Марфана, у пациентов отмечается краниосиностоз и задержка умственного развития. Может иметь место дилатация аорты.
Тактика ведения пациентов с синдромом Марфана
Патология зрения требует обследования глаз при рождении: обязательна биомикроскопия для определения эктопии хрусталика, по возможности, ранняя оптическая коррекция для предотвращения амблиопии. Показана консультация в специализированной офтальмологической клинике с определением тактики ведения и возможности хирургической коррекции вывиха или подвывиха хрусталика.
Большинство людей с синдромом Марфана близоруки, около 65% имеют косоглазие. Также широко распространены катаракта в среднем возрасте, глаукома и отслойка сетчатки. Последняя чаще встречается у детей и подростков, занимающихся групповыми игровыми видами спорта (баскетбол, волейбол, футбол, хоккей и другие), тяжелой атлетикой, акробатикой, аэробикой с прыжками. Эти разновидности физической культуры пациентам с синдромом Марфана противопоказаны. Заблаговременно должен быть обдуман выбор профессии. При планировании семьи и возможной беременности обязательно взвесить возможный риск. Семейный врач пациента должен быть координатором для исследований у соответствующих специалистов.
Патология сердечно–сосудистой системы является причиной смерти у 95% пациентов в молодом возрасте (до 40 лет). Особое внимание уделяется дилатации аорты с регургитацией, которая может прогрессировать годами и выявляться впервые к 18 годам. Наблюдение кардиолога предусматривает ежегодное проведение эходопплеркардиографии, а при расширении более чем на 50% от нормы – каждые полгода с обязательной консультацией кардиохирурга.
Аневризма аорты без лечения приводит к недостаточности аортального клапана или к расслоению аорты, сопровождающемуся сильными болями в области грудной клетки и ведущему к смерти 80% нелеченных пациентов в течение 14 дней. Наиболее яркий пример – случай со звездой американского волейбола, членом олимпийской сборной Flo Hyman, который умер от этого осложнения в 1986 г. Ранним признаком расслоения аневризмы восходящей аорты являются: охриплость голоса, боль и неприятные ощущения за грудиной, кашель, одышка, дисфагия или рвота, боли в спине.
В своевременной диагностике осложнений помогает параллельное мониторирование оксипролина и гликозаминогликанов суточной мочи, экскреция которых при резком прогрессировании дилатации значительно увеличивается (в 1,5–2–3 раза). Этот простой и дешевый метод можно рекомендовать как скрининговый для диагностики в динамике различных осложнений (прогрессирование недостаточности клапанов после инфекции и лихорадки, отслойка сетчатки, обострение хронического пиелонефрита, обострение остеоартроза, легочные осложнения – спонтанный пневмоторакс, пневмонию).
Диета пациентов с синдромом Марфана должна содержать достаточное количество магния, так как исследования, проведенные на лабораторных животных, показали, что при искусственном повреждении аорты баллоном и в последующем при диете с высоким содержанием магния идет более быстрое восстановление дефекта, чем при диетах с нормальным и низким содержанием магния. Кроме того, доказано, что назначение препаратов магния ведет к медикаментозной коррекции гиперкатехоламинемии и конституционального магниевого дефицита при пролапсе митрального клапана (ПМК).
Основой медикаментозного лечения является назначение b-адреноблокаторов. В случае расширенного корня аорты, и особенно при наличии регургитации они уменьшают выброс в аорту и соответственно нагрузку на стенки восходящего отдела, корригируют сопутствующую гемодинамическую артериальную гипертензию. Дозу у детей и взрослых увеличивают до тех пор, пока отношение периода предвыброса к периоду выброса не увеличится на 20%. Доза пропранолола может быть от 40 до 200 мг/сутки. Выгоднее использовать длительнодействующие b-адреноблокаторы, например, атенолол в дозе от 25 до 150 мг/сутки.
Регургитация при пролапсе митрального клапана, аритмии, симптомы гиперкатехоламинемии также требуют назначения b-адреноблокаторов. Считается, что b-адреноблокаторы предотвращают риск внезапной смерти у данной категории пациентов.
Однако в настоящее время показано, что b-адреноблокаторы при «чистом» ПМК приводят к ухудшению гемодинамики, что клинически проявляется усилением головокружения, слабости, снижением работоспособности, особенно у молодых лиц с гипотонией. Нами предложена методика по коррекции гемодинамики при ПМК с помощью венотонического препарата диосмин.
Хирургические методы лечения при синдроме Марфана имеют свои показания для протезирования: аневризматически расширенная аорта (более 6 см) заменяется эндо– или экзопротезом.
При пролапсе митрального клапана в случае «стабильной», даже выраженной регургитации, протезирование клапана не проводят, так как он имеет относительно благоприятное течение и прогноз в отсутствие осложнений. При быстром прогрессировании регургитации до выраженной или присоединении левожелудочковой недостаточности требуется замена митрального клапана.
У всех пациентов с патологией аорты и митрального клапана имеется риск инфекционного эндокардита, поэтому в случае малых и больших операционных вмешательств проводится первичная профилактика антибиотиками по установленной схеме.
Активность больных. Однозначно отрицательно решен вопрос о контактных, а также групповых, игровых видах спорта, изометрических нагрузках, работе с тяжестями. Показано плавание. Положительное влияние дозированных физических нагрузок на состояние кардио–респираторной системы у пациентов с дисплазиями соединительной ткани еще нуждается в обосновании при синдроме Марфана.
Скелетопатии при синдроме Марфана обычно выявляются к возрасту 5 лет и иногда прогрессируют с невероятной быстротой, хотя удлиненные размеры конечностей и пальцев – долихостеномелия и арахнодактилия заметны уже у новорожденного. Есть указания, что в подобных случаях виновен дефицит некоторых макроэлементов (кальций, магний, цинк, медь) и белков, участвующих в «строительстве» соединительной ткани. Большое место в зарубежной литературе занимают сообщения об эффективности пищевых добавок, содержащих вышеперечисленные макроэлементы, а также гиалуроновую кислоту, викасол, колекальциферол.
В крови больных нередко отмечается повышенный уровень соматотропного гормона, в связи с чем для «подавления избыточного роста» рекомендуются высокожировые энпиты в питании с раннего детства, уменьшающие секрецию соматотропного гормона.
Оперативное лечение деформаций грудной клетки и позвоночника является травматичной процедурой, нередко осложняющейся плевритами, перикардитами, пневмониями в раннем и позднем послеоперационных периодах. Вопрос о целесообразности торакопластики при деформациях грудной клетки многократно обсуждался на симпозиумах, посвященных дисплазиям соединительной ткани, и специалисты различных регионов Сибири приняли позицию, отрицающую целесообразность торакопластики при синдроме Марфана. В зарубежной литературе показанием к торокопластике считаются большие степени воронкообразной деформации – 3 и 4 степени.
Сравнение показателей гемодинамики до и после оперативного вмешательства по поводу деформаций грудной клетки у лиц, прооперированных в г. Омске, показало, что ожидаемого улучшения гемодинамики в позднем послеоперационном периоде не получено, несмотря на некоторые позитивные сдвиги в раннем послеоперационном периоде. Это еще раз подтверждает мнение о том, что выявляемые кардио–респираторные нарушения при дисплазиях соединительной ткани и синдроме Марфана, как частном ее проявлении, в большей степени связаны с собственно органообразующим дефектом соединительной ткани, чем с механическим влиянием деформированной грудной клетки на внутриторакальные органы.
Примерная схема метаболической терапии (модифицировано по Кадуриной Т.И. ) с преимущественным воздействием на соединительную ткань при синдроме Марфана должна включать аскорбиновую кислоту (при отсутствии оксалурии и семейного анамнеза по мочекаменной патологии) в виде коктейлей с молоком, йогуртом; доза – от 1,0 до 4,0 г в день в зависимости от возраста.
Рекомендуются препараты, содержащие глюкозаминсульфат; доза в возрасте 12 лет и взрослых – по 1,5 г 1 раз в день во время еды, запивать большим количеством воды; курс – 1,5 месяца. В следующем курсе можно использовать препараты, содержащие хондроитинсульфат. Доза: детям в возрасте до 1 года – 250 мг, от 1 года до 5 лет – 500 мг, от 6 до 12 лет – 500–750 мг, взрослым – 1,5–2,0 г во время еды; запивать большим количеством воды; курс – 2 месяца.
В качестве метаболита в курс можно включить янтарную кислоту (в капсуле 100 мг); по 1–2 капсулы 2 раза в день, курс – 3 недели. Широко используются препараты магния.
Карнитина хлорид, 20 % раствор; доза детям в возрасте 1 год – по 5–10 капель, от 1 года до 6 лет – по 15 капель, от 6 до 12 лет – по 30–40 капель, старше 12 лет – по 1 чайной ложке 3 раза в день, после еды; курс – 1 месяц. В терапии активно рекомендуются витаминно–минеральные комплексы курс – 1 месяц; добавки, содержащие L–лизин; доза – в зависимости от возраста; кратность приема – 2 раза в день; курс – 1 месяц; токоферол; доза в возрасте 12 лет и взрослым от 400 до 800 МЕ в сут.; курс – 3 недели.
Беременность при синдроме Марфана
Беременность при синдроме Марфана опасна, по крайней мере, по двум причинам. Во–первых, имеется риск наследования, который составляет 50% по закону Менделя. Во–вторых, во время третьего триместра беременности и в раннем послеродовом периоде резко увеличивается риск формирования аневризмы аорты, разрыва, расслоения уже существующей аневризмы аорты и возникновения септического эндокардита. Расслоение аневризмы аорты во время беременности связано с увеличением объема циркулирующей крови, аорто–кавальной компрессией и гормональными изменениями. Риск развития этого осложнения растет пропорционально увеличению срока беременности.
Для диагностики аневризмы аорты во время беременности наиболее информативно использовать эхокардиографию (ЭхоКГ), позволяющую определить размеры аорты несколько раз во время беременности, перед родами и в послеродовом периоде. Всем беременным с синдромом Марфана профилактически назначают b-адреноблокаторы, уменьшающие сердечный выброс и прогрессирование дилатации аорты с риском ее расслоения.
Роды естественным путем возможны у женщин без выраженной патологии сердечно–сосудистой системы, и при диаметре аорты, не превышающем 4 см. Беременность и роды у таких женщин проходят, как правило, без серьезных осложнений для матери и плода.
Методом выбора при обезболивании родов у рожениц с синдромом Марфана следует считать эпидуральную анальгезию растворами местных анестетиков низкой концентрации с добавлением наркотических анальгетиков. Обезболивание должно начинаться с первого периода родов. В связи с этим пункцию эпидурального пространства следует выполнить с самого начала родовой деятельности. В первом периоде родов проводят эпидуральную анальгезию местными анестетиками (лидокаин 0,5 % или бупивакаин 0,75%) с добавлением наркотических анальгетиков или агонист–антагонистов. Предпочтительно проведение постоянной эпидуральной инфузионной анальгезии, которая характеризуется минимальными изменениями гемодинамики, в основном с уменьшением ударного выброса сердца в связи с частичным депонированием крови в дилатированных сосудах нижних конечностей. Если по каким–то причинам к началу потужного периода анальгезия оказывается недостаточной, необходимо полностью исключить потуги наложением акушерских щипцов.
Когда диаметр корня аорты превышает 5,5 см, необходимо досрочное родоразрешение, даже при отсутствии каких–либо субъективных симптомов. Существует мнение, что у таких женщин следовало бы производить хирургическую коррекцию аневризмы аорты во время беременности даже при отсутствии каких–либо угрожающих симптомов, поскольку при дилатированной аорте они могут появиться в любой момент.
Предоперационная подготовка перед плановой операцией кесарева сечения должна включать назначение b-адреноблокаторов и вазодилататоров. Методом выбора обезболивания при оперативном родоразрешении является эпи– или субдуральная анестезия.
Следует помнить, что риск расслоения аневризмы аорты сохраняется в течение нескольких недель после родов, так как в раннем послеродовом периоде за счет декомпрессии нижней полой вены и перераспределения крови может резко возрасти сердечный выброс.
Одной из причин смерти родильниц с синдромом Марфана может быть септический эндокардит. В связи с этим в послеродовом или в послеоперационном периоде необходима антибактериальная терапия.
В случае клинической смерти роженицы на фоне неэффективного закрытого массажа сердца не уточняется, надо ли предпринимать торакотомию и прямой массаж сердца. Но операцию кесарева сечения на фоне реанимационных мероприятий при жизнеспособном плоде безусловно следует проводить.
В литературе встречаются сообщения о проведении операции кесарева сечения после остановки кровообращения, когда реанимация оказалась неэффективной. Интерес представляет не сам факт операции, а то, что сразу же после извлечения плода удавалось восстановить сердечную деятельность матери. Быстро предпринятое оперативное родоразрешение значительно увеличивает вероятность успеха проводимых реанимационных мероприятий. Операция кесарева сечения, выполненная в агональном состоянии или даже после остановки кровообращения, позволяет в такой ситуации считать ее составной частью реанимационных мероприятий.
Большинство детей, извлеченных в такой драматической ситуации, выживают, если операция произведена в течение 5 мин. после остановки кровообращения и даже через 21 минуту после остановки кровообращения, когда реанимационный комплекс не сопровождался признаками адекватного мозгового кровотока. Полагают, что при проведении сердечно–легочной реанимации у беременных в поздних сроках беременности при синдроме Марфана операция кесарева сечения должна выполняться как можно раньше и считаться одним из компонентов комплекса реанимационных мероприятий.
Таким образом, спектр патологии при синдроме Марфана затрагивает многие органы и системы, поэтому в идеале лечение осуществляется группой врачей разных специальностей с координацией действий врачом общей практики (семейным врачом). Синдром Марфана – нечастая патология в популяции (1:10000), но требует от врачей разных профилей конкретных знаний по диагностике и профилактическим мероприятиям, являющихся залогом продления жизни пациентам.
| Эти глаза напротив | ОФТАЛЬМОЛОГИЯ | Пятница, 14 декабря, 2018г. |
| Светобоязнь, фотокератит и солнечные очки | ОФТАЛЬМОЛОГИЯ | Пятница, 14 декабря, 2018г. |
| Отёки | РАЗНОЕ + ЭТО ИНТЕРЕСНО | Среда, 4 ноября, 2015г. |
Вы здесь » В ГОСТЯХ У ОЛЬГИ-АЛЁНУШКИ » РАЗНОЕ + ЭТО ИНТЕРЕСНО » Врождённые и генетические заболевания

